#Origine du #SARS-CoV-2 et du #Covid19 : le point sur l’enquête en cours et les dernières hypothèses
Virginie Courtier, Université Paris Cité et Etienne Decroly, Aix-Marseille Université (AMU)
En mars 2021, la commission conjointe Chine-OMS qui s’était rendue à Wuhan concluait dans son rapport que l’épidémie de Covid-19 avait très probablement une origine zoonotique (animale) et que l’hypothèse d’un accident de laboratoire était très improbable… Cependant, à cette même occasion, le directeur général de l’OMS, Tedros Adhanom Ghebreyesus, prenait à contre-pied cette annonce en affirmant que toutes les hypothèses restaient ouvertes et qu’il restait des questions qui « devront être traitées dans le cadre d’études supplémentaires ».
Plus de deux ans et demi se sont désormais écoulés depuis les premiers cas détectés en Chine, en décembre 2019. Quelles sont aujourd’hui les dernières avancées concernant l’origine du SARS-CoV-2, le virus responsable du Covid-19 ? Avons-nous fait les progrès nécessaires depuis notre dernière analyse sur le sujet pour résoudre cette question brûlante ? L’enjeu est de déterminer l’origine de cette pandémie afin d’essayer d’éviter de nouvelles épidémies similaires dans le futur.
À ce jour, ni les virus progéniteurs (ancestraux) de l’épidémie ni l’animal responsable des premières transmissions du virus à l’espèce humaine n’ont été identifiés.
En absence de ces éléments, différentes hypothèses restent d’actualité : une origine zoonotique à partir de virus circulant dans des chauves-souris (en passant ou non par des hôtes intermédiaires), une infection dans une grotte associée ou non à la recherche (tourisme, prélèvements, etc.), ou bien une infection par un virus stocké voire manipulé génétiquement dans un laboratoire à Wuhan.
Les difficultés d’une telle enquête
Reconstruire les chaînes de transmission à l’origine des épidémies est un travail difficile, et il faut être conscient qu’il restera toujours des zones d’ombre. Par exemple, pour une épidémie donnée, l’identification du « patient zéro », c’est-à-dire du premier humain infecté, est souvent difficile, voire impossible. Néanmoins, on peut espérer obtenir des échantillons des virus circulant au début d’une épidémie.
Ces informations, complétées par des données épidémiologiques (sérologie, séquences des virus, test PCR, etc.), permettent d’élaborer divers scénarios probables.
Dans le cas du SARS-CoV-2, le travail est d’autant plus ardu que le virus ne reste que quelques semaines dans l’organisme infecté (contrairement au virus du sida qui est intégré dans le génome des cellules infectées) et que de nombreuses personnes sont asymptomatiques. Sans compter le fait que ce sujet est sensible…
Dans certains pays comme la Chine ou les États-Unis, il est difficile d’obtenir des informations, notamment sur les virus collectés ou sur les recherches menées dans les laboratoires chinois au moment de l’émergence de l’épidémie.
Dans la suite de cet article, nous présentons et discutons les principaux éléments nouveaux sur le sujet.
Les données apportées par la génétique
Deux études publiées dans la revue Science en juillet 2022 argumentent que l’épidémie aurait commencé au marché de Huanan, à Wuhan.
La première examine la localisation géographique des premiers cas répertoriés de Covid-19 et montre leur regroupement autour de ce marché. Cette étude indique également que les premiers cas humains identifiés sont concentrés dans la zone du marché fréquentée par les vendeurs d’animaux sauvages vivants potentiellement infectables par le SARS-CoV-2 – tels que le renard roux, le chien viverrin ou le blaireau. De plus, des prélèvements environnementaux réalisés dans cette zone se sont révélés positifs au SARS-CoV-2.
La seconde étude, basée sur l’analyse des séquences des premiers virus SARS-CoV-2, indique que deux lignées virales distinctes notées A et B circulaient au début de l’épidémie. Les premières infections humaines (entre le 23 octobre et le 8 décembre 2019) impliqueraient la lignée B alors que le virus de la lignée A aurait franchi la barrière d’espèce quelques semaines plus tard.

Arend Kuester/Flickr, CC BY-NC
De ces deux analyses, les auteurs concluent qu’au moins deux franchissements de la barrière d’espèce ont eu lieu, ce qui est une caractéristique attendue des transmissions zoonotiques naturelles, et que le virus serait passé des animaux aux humains via des animaux vendus au marché.
Toutefois, aucun animal hôte intermédiaire portant le virus progéniteur n’a été identifié sur place. Les prélèvements positifs du marché semblent d’origine humaine puisqu’ils présentent les mêmes séquences virales que celles retrouvées chez les patients. De plus, les premiers cas occupaient des étals trop dispersés pour imaginer une contamination directe à partir d’une ou deux sources animales.
Il est intéressant également de noter que les premiers patients détectés infectés par la lignée A n’ont pas révélé de lien épidémiologique avec des marchés.
Deux mutations seulement distinguent la lignée A de la lignée B. La lignée A est plus proche des virus de chauves-souris, et serait apparue avant la lignée B. Comme deux mutations peuvent parfois apparaître en une seule transmission interhumaine, on peut faire l’hypothèse que ces deux mutations sont apparues au sein des humains et non des animaux sauvages du marché.
De plus, il y a probablement eu des biais d’échantillonnage, dans la mesure où les premières recommandations des autorités chinoises étaient de ne séquencer que les virus provenant de patients ayant fréquenté le marché.
En résumé, ces deux études montrent que le marché a constitué un épicentre précoce de la pandémie, dans le sens où il a joué un rôle amplificateur de l’épidémie. Mais est-ce le site où l’épidémie a commencé ?
En fait, les événements en amont du marché, ainsi que les circonstances qui ont favorisé la propagation du virus au marché, restent un mystère.
Certains auteurs de ces articles ont pu avoir des propos très optimistes dans la presse quand ils ont affirmé que leurs travaux écartaient définitivement l’hypothèse de l’accident de laboratoire. En réalité, des études supplémentaires doivent être poursuivies pour élucider l’origine de l’épidémie.
[Plus de 80 000 lecteurs font confiance à la newsletter de The Conversation pour mieux comprendre les grands enjeux du monde. Abonnez-vous aujourd’hui]
À la recherche des premières séquences et des premiers cas
Retracer les origines d’une épidémie suppose de remonter les chaînes de transmission afin d’identifier la séquence des premiers virus humains et des virus progéniteurs. Ceci pour comprendre comment les premiers humains ont été contaminés.
Malheureusement, l’accès aux données par la communauté scientifique reste problématique. Malgré des demandes répétées, les experts de l’OMS n’ont toujours pas eu accès aux échantillons biologiques du début de l’épidémie détenus par la Chine.
Malgré ces difficultés, des séquences virales associées au début de l’épidémie ont pu être exhumées de bases de données.
Les séquences présentes dans les bases de données peuvent provenir non seulement de l’espèce que les chercheurs avaient initialement l’intention d’étudier, mais aussi du matériel génétique environnant – présent soit au moment de la collecte, soit apporté a posteriori suite à une contamination lors du traitement ultérieur des échantillons en laboratoire.
Ainsi, en analysant les données brutes de séquençage d’échantillons de sols collectés en Antarctique, un variant apparemment précoce du SARS-CoV-2 et proche de la lignée A a été identifié. Or ces échantillons de sol n’ont rien à voir avec le Covid-19… Ce variant du SARS-CoV-2 identifié aurait contaminé les échantillons lors du séquençage en Chine, entre décembre 2019 et janvier 2021. Son origine reste mystérieuse, car il possède quelques mutations introuvables au début de la pandémie (dont une délétion de 27 paires de bases).
D’autre part, le virologue Jesse Bloom, en utilisant des archives du web, a retrouvé la séquence partielle de plusieurs virus SARS-CoV-2 obtenus sur des patients du début de l’épidémie en Chine. Ces séquences avaient été supprimées des bases de données du NCBI (National Center for Biotechnology Information), suite à la demande des chercheurs chinois qui les avaient déposées – ils disposaient du droit de les retirer à tout moment. Ce travail doit se poursuivre dans la mesure où d’autres séquences de SARS-CoV-2 ont été retirées des bases de données du NCBI, à la demande de divers auteurs basés en Chine et aux États-Unis.
Enfin, la recherche des premiers cas de contamination se poursuit, avec l’analyse des échantillons de patients européens dans la période prépandémique, c’est-à-dire fin 2019. Deux études italiennes récentes suggèrent par exemple que le virus circulait déjà à l’automne dans le nord de la péninsule. Malheureusement, la séquence complète de ces virus n’a pas pu être obtenue. Il est donc impossible de conclure avec certitude s’ils correspondent à la lignée A, B ou une autre… Il est toujours possible d’imaginer que ces cas résultent d’infections par d’autres coronavirus ou de contaminations ultérieures des échantillons lors de leur manipulation.
Pour avoir des preuves irréfutables de la présence du virus avant décembre 2019, il faudrait obtenir des séquences virales complètes présentant des séquences intermédiaires qui n’ont jamais été séquencées auparavant. Pour l’instant, les échantillons positifs se sont avérés trop dégradés pour y parvenir.
Quel réservoir pour le virus, et quel progéniteur pour l’épidémie ?
Une approche alternative consiste à rechercher dans les réservoirs naturels des virus parents ou cousins proches du SARS-CoV-2.
Le virus le plus proche identifié au début de l’épidémie était RaTG13, provenant d’une mine du Yunnan à 1500 km au sud de Wuhan et séquencé par l’institut de virologie de Wuhan. Plus de 1000 mutations le séparent du SARS-CoV-2. Ces mutations indiquent que RaTG13 est un cousin de SARS-CoV-2 et que leur virus-ancêtre circulait dans les chauves-souris il y a 30-50 ans. Mais RaTG13 n’est pas le progéniteur de l’épidémie, et il n’est pas capable de reconnaître efficacement les récepteurs ACE2 présents à la surface des cellules humaines, limitant de facto la possibilité d’infection humaine.
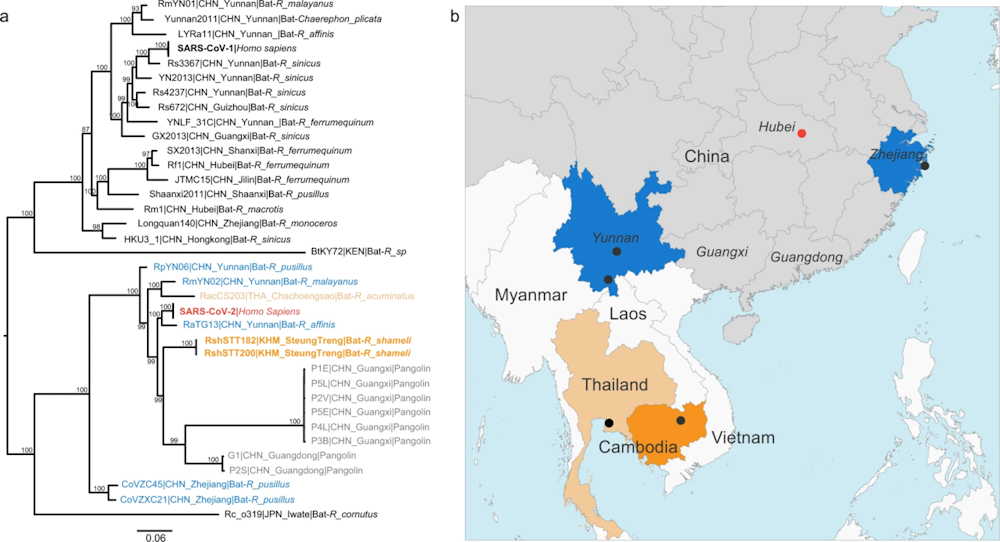
Nature, d’après A novel SARS-CoV-2 related coronavirus in bats from Cambodia, D. Delaune and al, CC BY
Ces deux dernières années, le laboratoire qui a obtenu le plus de données importantes sur la biologie des virus pour comprendre l’origine de SARS-CoV-2 est l’Institut Pasteur à Paris.
En effectuant des collectes au Laos en 2020, l’Institut Pasteur a identifié dans des populations de chauves-souris de nouveaux cousins plus proches du SARS-CoV-2 que RaTG13. Ces virus contiennent un domaine de reconnaissance du récepteur ACE2 qui ressemble beaucoup à celui de SARS-CoV-2 et qui leur permet cette fois d’infecter des cellules portant le récepteur ACE2 humain.
Cette découverte suggère que des virus de chauve-souris pourraient franchir la barrière d’espèce sans nécessité d’hôte intermédiaire. Toutefois, l’infection des cellules humaines et des modèles murins et simiens est limitée, car les virus laotiens ne contiennent pas de site de clivage par la furine. Or ce dernier, présent chez le SARS-CoV-2, joue un rôle capital dans le tropisme pulmonaire de l’infection et la propagation interhumaine du virus. Il aurait pu être sélectionné au cours de l’évolution, car il rend le virus plus transmissible.
Pour tester cette hypothèse, les équipes de l’Institut Pasteur ont réalisé des expériences d’évolution virale par passages successifs du virus dans des souris transgéniques portant le récepteur ACE2 humain et dans des cellules humaines. Le site de clivage par la furine n’est pas apparu, malgré six infections consécutives de souris. Ces résultats suggèrent que l’acquisition du site de clivage par la furine, pendant la phase précoce de circulation silencieuse d’un virus de type Laotien au sein de la population humaine, est peu probable, d’autant que l’analyse de populations humaines très exposées à ces virus n’a pas permis d’identifier d’infection.
Le plus probable est que ce site furine existe chez des virus de chauve-souris actuellement non identifiés, ou bien qu’il soit apparu naturellement dans un animal suite à une recombinaison avec un virus possédant ce site de clivage… ou lors de manipulations génétiques visant à l’ajouter.
La découverte de virus infectant les chauves-souris qui seraient encore plus proches de SARS-CoV-2, ou de sarbécovirus ayant un site de clivage par la furine, permettrait de départager ces hypothèses.
La question des recherches dans les laboratoires de Wuhan
À Wuhan, les recherches sur les virus des chauves-souris sont menées sur trois sites :
- à Zhengdian (sud de Wuhan), où se trouve le laboratoire P4 de l’institut de virologie de Wuhan (Wuhan Institute of Virology, WIV),
- à Xiaohongshan, qui est l’emplacement historique du WIV,
- et au Center for Disease Control (CDC), installé à quelques minutes du marché de Huanan, depuis décembre 2019.
Ces laboratoires stockent et étudient des virus prélevés en Asie sur divers animaux sauvages – sachant que la Chine a lancé un programme national pour répertorier tous ces virus en 2019. Une personne a pu être contaminée par accident en manipulant des échantillons infectés.
Même si la séquence de SARS-CoV-2 ne correspond à aucun virus qui avait été déjà publié auparavant par un laboratoire, et que le site de clivage par la furine n’est pas celui que les chercheurs utilisent habituellement, on peut se demander si des manipulations génétiques en cours auraient pu conduire au SARS-CoV-2.
En septembre 2021, un document rédigé en 2018 a été divulgué par DRASTIC, un collectif de personnes issues de divers horizons (bactériologie, biotechnologies, neurobiologie, ingénierie, science des données, etc.) et réunies sur Twitter qui s’est donné pour mission d’explorer les origines du SARS-CoV-2.
Ce document révèle que des expériences d’insertion de divers sites de clivage à la furine dans des virus de chauves-souris avaient été imaginées en collaboration entre le WIV et des laboratoires américains. Le projet, soumis à la DARPA, n’a pas été financé et nous ne savons pas si de telles expériences ont finalement été menées.
Enfin, la capacité du WIV à construire des virus chimériques pour étudier les mécanismes de franchissement de barrière d’espèce est maintenant avérée. Ce type d’expérience a en effet été réalisé avec succès sur le SARS-CoV dès 2017. De plus, un rapport intermédiaire d’activité, obtenu sous contrainte judiciaire (par le Freedom of Information Act, FOIA), confirme que des virus chimériques MERS-CoV (expérience dite de gain de fonction) ont été construits en Chine – le MERS étant aussi un coronavirus, mais relativement éloigné du SARS-CoV-2, qui a émergé en 2012 et provoque des infections aiguës.
Ce genre de manipulation génétique ne laisse généralement pas de traces dans la séquence.
L’hypothèse d’une création et d’une contamination volontaire nous semble improbable pour deux raisons : tous les pays ont été pris au dépourvu par l’arrivée des premiers cas de Covid-19, et aucun antidote n’était disponible au début de la pandémie.
Les derniers avis en date de deux commissions internationales
En août 2021, l’OMS a mis en place un nouveau Groupe consultatif scientifique sur les origines des nouveaux agents pathogènes (Scientific Advisory Group for the Origins of Novel Pathogens, SAGO). Le SAGO comprend une vingtaine d’experts internationaux et l’une de ses missions est d’orienter l’OMS sur les prochaines mesures à prendre pour comprendre quelles sont les origines du SARS-CoV-2.
En parallèle, le journal scientifique The Lancet a assemblé une commission interdisciplinaire pour tirer des enseignements de la crise Covid-19 pour l’avenir et examiner l’origine de l’épidémie.
Le SAGO a rendu public son premier rapport en juin 2022, tandis que la commission _Lancet_a publié son rapport final le 14 septembre.
Les deux concluent que l’origine du coronavirus n’est pas encore établie et que plusieurs hypothèses doivent être envisagées :
- une transmission zoonotique d’un animal à un humain non liée à la recherche,
- une infection à la suite d’activités liées à la recherche, avec trois voies possibles : une infection sur le terrain lors de la collecte d’échantillons, une infection en laboratoire lors de l’étude de virus non modifiés ou une infection par un virus manipulé génétiquement.
Que peut-on conclure, et où faut-il chercher ?
Jusqu’à présent, aucune enquête détaillée indépendante, internationale, transparente et scientifique n’a été menée sur la possibilité d’un accident de laboratoire. Les carnets de laboratoire, les courriers électroniques, les bases de données et les échantillons des institutions impliquées dans ces recherches n’ont pas été mis à la disposition des chercheurs indépendants ou de l’OMS par la Chine.
De nombreuses pistes s’offrent encore aux experts pour essayer de comprendre comment cette pandémie a commencé :
Sur 100 cas confirmés par PCR de Covid-19 en Chine avec symptômes en décembre 2019, seule une vingtaine de génomes sont actuellement disponibles. Il faudrait avoir accès aux génomes de ces autres patients ;
Continuer les recherches de cas précoces, en Europe et ailleurs ;
- Pouvoir analyser plus en profondeur les séquences des bases de données, notamment celles qui ont été supprimées
- Récupérer la base de données des virus de WIV qui a été retirée d’Internet et autres documents de laboratoire ;
- Renforcer la surveillance, dans l’hypothèse de trouver de nouveaux épisodes de franchissement de la barrière d’espèces vers les humains, avec de nouveaux variants de SARS-CoV-2, comme avec le MERS ou le virus du sida. Il faudra alors être très réactif pour essayer de retrouver l’hôte intermédiaire.
- Poursuivre la recherche de coronavirus proches chez les chauves-souris de l’écosystème où ont été trouvés des précurseurs proches de SARS-CoV-2.

Virginie Courtier, Directrice de recherche CNRS, génétique et évolution, Université Paris Cité et Etienne Decroly, Directeur de recherche en virologie, Aix-Marseille Université (AMU)
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original.